Code
PsychCore Genomics Pipeline
This containerized pipeline was developed for high-throughput parallel processing on the Amazon Web Services cloud platform. It was deployed to process whole-genome sequencing data from FASTQ to VCF for analysis of the human prefrontal cortex across development.
MagellanMapper
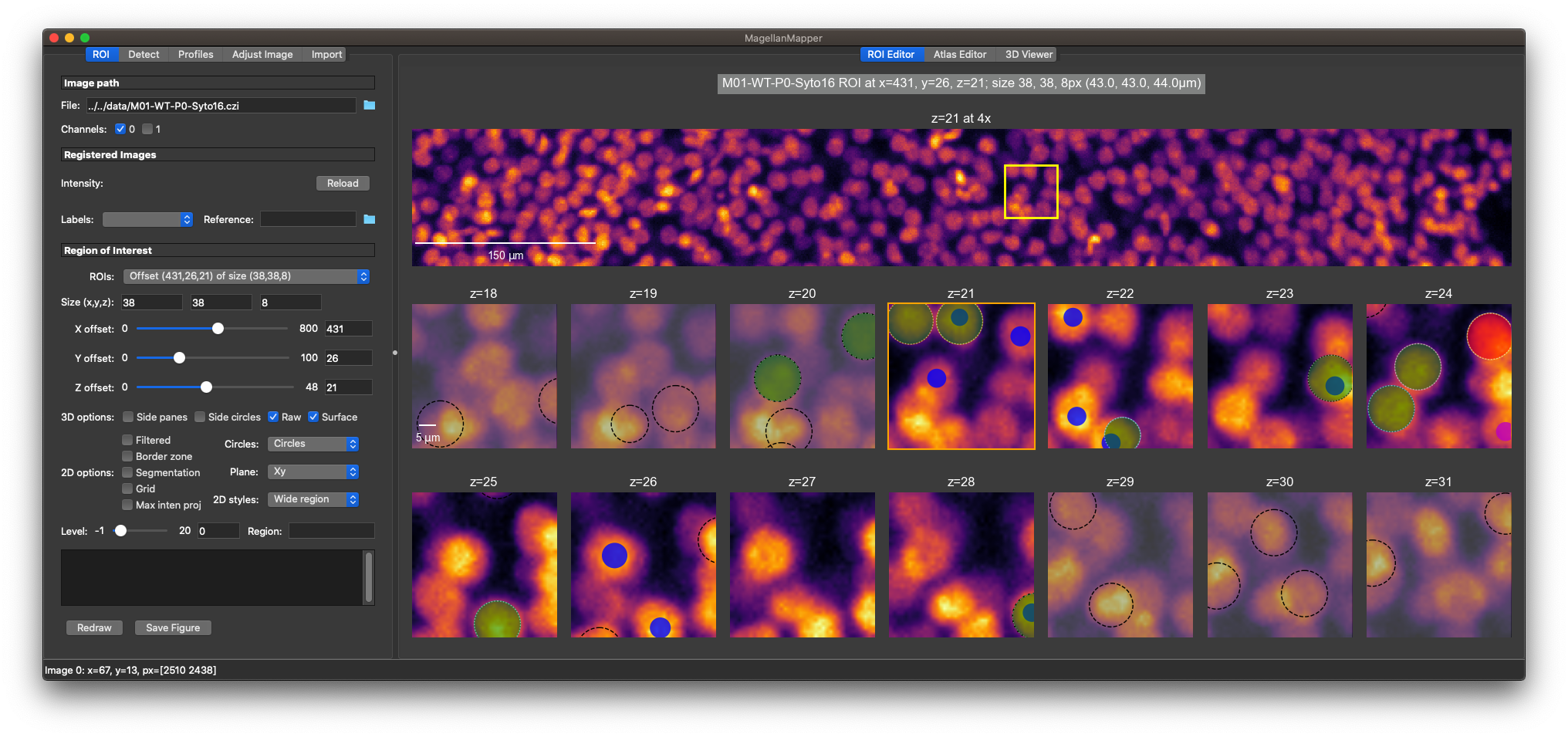
MagellanMapper is a graphical imaging informatics suite and pipeline for 3D reconstruction and automated analysis of and whole specimens and atlases. Its design philosophy is to make the raw 3D images as accessible as possible, simplify annotation from nuclei to atlases, and scale from the laptop or desktop to the cloud in cross-platform environments.
wgsPowerTest
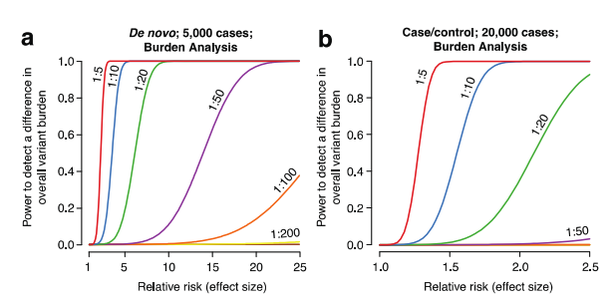
This R package runs power calculations for the discovery of variants in whole genome sequencing data.
SCN2A Variant Browser
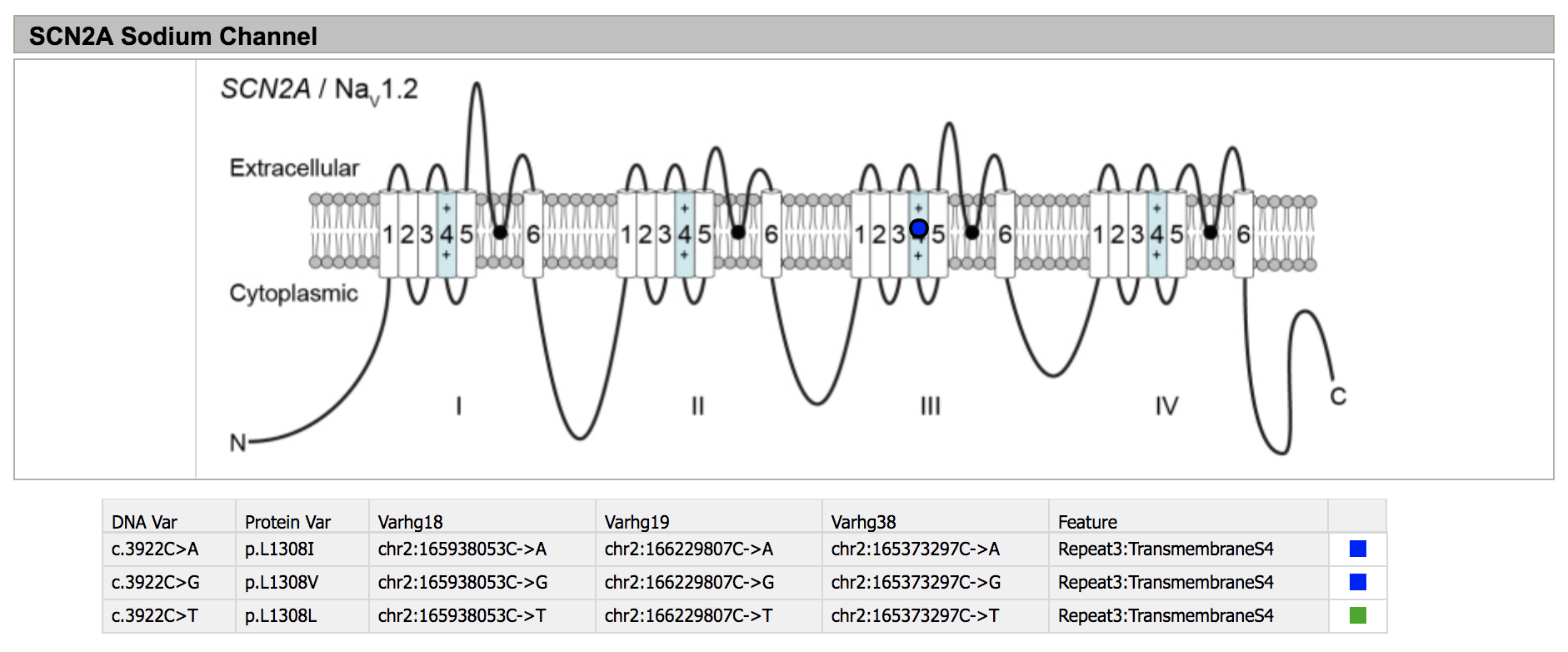
The database SCN2A variants.
CNVision
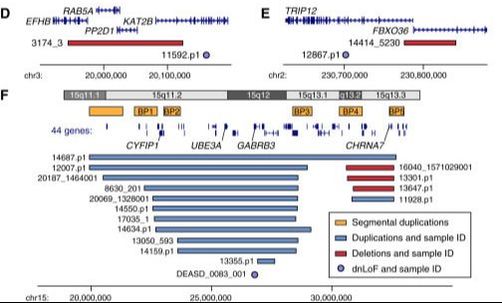
CNVision is designed for detecting and scoring Copy Number Variants (CNVs) from Illumina SNP genotyping data. It runs in a UNIX environment and works with all Illumina chips (from 300k to latest Omni). CNVs are predicted using PennCNV, QuantiSNPv2.3, and GNOSIS (an in-built algorithm). The predicted CNVs are merged, joined (if appropriate), and scored based on the per SNP variability in the raw genotyping data. CNVision can also identify de novo CNVs in family-based data using the per SNP variability algorithm. Comparison with 1000 Genomes, the Genome Structural Variation Consortium, and replicate Illumina data demonstrates the efficacy of the CNV scoring method in both inherited and de novo CNVs. CNVision was written to analyze data for the Simons Simplex Collection autism data. A full description of methods are given in the following paper which can be used to reference (Sanders et al. (2015))
Identity check
Managing large genomic datasets requires accurate estimation of sample identity. This script rapidly identifies all BAM files and Illumina SNP genotyping FinalReports on a cluster, generates a SNP barcode from each one, and uses BLAT to identify duplicates and/or matches. It is run off aligned, indexed BAM files directly (hg18 or hg19) and FinalReports directly (hg18 or hg19). Cross platform (BAM to FinalReport) and cross genome build (hg18 to hg19) is handled automatically.
UNIX treasure hunt tutorial
This perl script will install a series of directories and clues that teaches basic UNIX command line skills including cd, ls, grep, less, head, tail, and nano. Run the perl script from the command line on a UNIX based machine (e.g. Mac or Linux) using the command: perl treasureHunt_v2.pl. Then use ls to find the first clue. A PDF of command line commands is also available to download.